
AT7867 dihydrochloride,99.17%
产品编号:Bellancom-12059A| CAS NO:1431697-86-7| 分子式:C20H22Cl3N3| 分子量:410.77
本网站销售的所有产品仅用于工业应用或者科学研究等非医疗目的,不可用于人类或动物的临床诊断或者治疗,非药用,非食用,
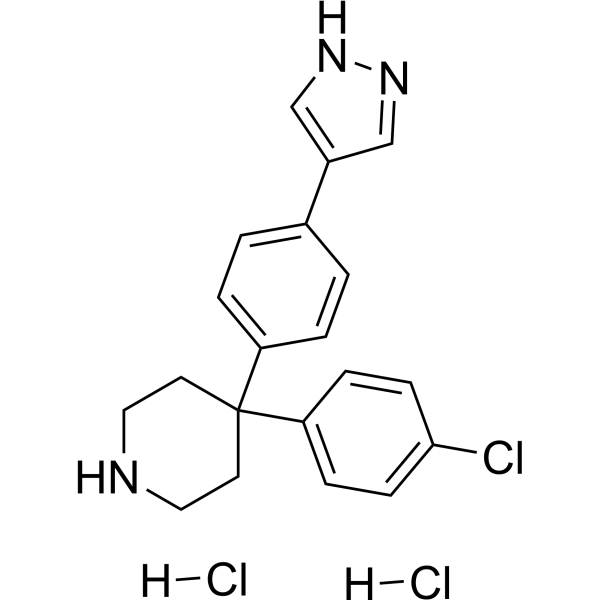
AT7867 dihydrochloride
| 产品介绍 | AT7867 dihydrochloride 是一种 ATP 竞争性的 Akt1/Akt2/Akt3 和 p70S6K/PKA 抑制剂,IC50 分别为 32 nM/17 nM/47 nM 和 85 nM/20 nM。 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 生物活性 | AT7867 dihydrochloride is a potent ATP-competitive inhibitor of Akt1/Akt2/Akt3 and p70S6K/PKA with IC50s of 32 nM/17 nM/47 nM and 85 nM/20 nM, respectively. | ||||||||||||||||
| 体外研究 |
The inhibition of AKT2 by AT7867 is shown to be ATP-competitive with a Ki of 18nM. AT7867 also displays potent activity against the structurally related AGC kinases p70S6K and PKA, but shows a clear window of selectivity against kinases from other kinase sub-families. In vitro growth inhibition studies show that AT7867 blocks proliferation in a number of human cancer cell lines. AT7867 appears to be most potent at inhibiting proliferation in MES-SA uterine, MDA-MB-468 and MCF-7 breast, and HCT116 and HT29 colon lines (IC50 values range from 0.9-3 μM), and least effective in the two prostate lines tested (IC50 values range from 10-12 μM) . 西域 has not independently confirmed the accuracy of these methods. They are for reference only. | ||||||||||||||||
| 体内研究 |
Following oral administration at 20 mg/kg, the elimination of AT7867 from plasma appears to be similar to that observed after i.v. administration. Plasma levels of AT7867 remain above 0.5 μM for at least 6 hours following an oral dose of 20 mg/kg. Assuming linear pharmacokinetics following i.v. administration, the bioavailability by the oral route is calculated to be 44%. In vivo pharmacodynamic (PD) biomarker studies are therefore performed with this model. Following pharmacokinetic and tolerability studies, doses of AT7867 (90 mg/kg p.o. or 20 mg/kg i.p.) are administered to athymic mice bearing MES-SA tumors and the phosphorylation status of GSK3β and S6RP in tumors is monitored over time. Clear inhibition of phosphorylation of the two markers of pathway activity is seen at 2 and 6 hours following treatment with AT7867. By 24 hours, total levels of both GSK3β and S6RP are greatly reduced. 西域 has not independently confirmed the accuracy of these methods. They are for reference only. | ||||||||||||||||
| 体内研究 |
Following oral administration at 20 mg/kg, the elimination of AT7867 from plasma appears to be similar to that observed after i.v. administration. Plasma levels of AT7867 remain above 0.5 μM for at least 6 hours following an oral dose of 20 mg/kg. Assuming linear pharmacokinetics following i.v. administration, the bioavailability by the oral route is calculated to be 44%. In vivo pharmacodynamic (PD) biomarker studies are therefore performed with this model. Following pharmacokinetic and tolerability studies, doses of AT7867 (90 mg/kg p.o. or 20 mg/kg i.p.) are administered to athymic mice bearing MES-SA tumors and the phosphorylation status of GSK3β and S6RP in tumors is monitored over time. Clear inhibition of phosphorylation of the two markers of pathway activity is seen at 2 and 6 hours following treatment with AT7867. By 24 hours, total levels of both GSK3β and S6RP are greatly reduced. 西域 has not independently confirmed the accuracy of these methods. They are for reference only. | ||||||||||||||||
| 性状 | Solid | ||||||||||||||||
| 溶解性数据 |
In Vitro:
DMSO : 50 mg/mL (121.72 mM; Need ultrasonic) H2O : 2.27 mg/mL (5.53 mM; ultrasonic and warming and heat to 60°C) 配制储备液
*
请根据产品在不同溶剂中的溶解度选择合适的溶剂配制储备液;一旦配成溶液,请分装保存,避免反复冻融造成的产品失效。 In Vivo:
请根据您的实验动物和给药方式选择适当的溶解方案。以下溶解方案都请先按照 In Vitro 方式配制澄清的储备液,再依次添加助溶剂:
——为保证实验结果的可靠性,澄清的储备液可以根据储存条件,适当保存;体内实验的工作液,建议您现用现配,当天使用;
以下溶剂前显示的百
| ||||||||||||||||
| 运输条件 | Room temperature in continental US; may vary elsewhere. | ||||||||||||||||
| 储存方式 |
4°C, sealed storage, away from moisture *In solvent : -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture) | ||||||||||||||||
| 参考文献 |





 浙公网安备 33010802013016号
浙公网安备 33010802013016号