
Teglicar 替格列卡,98.0%
产品编号:Bellancom-16482| CAS NO:250694-07-6| 分子式:C22H45N3O3| 分子量:399.61
Teglicar 是选择性和可逆性的肉毒碱棕榈酰转移酶 1 (L-CPT1) 肝异构体抑制剂,可减少酮生成和葡萄糖生成,降低糖异生,改善葡萄糖稳态。Teglicar 具有潜在的抗高血糖特性。
本网站销售的所有产品仅用于工业应用或者科学研究等非医疗目的,不可用于人类或动物的临床诊断或者治疗,非药用,非食用,
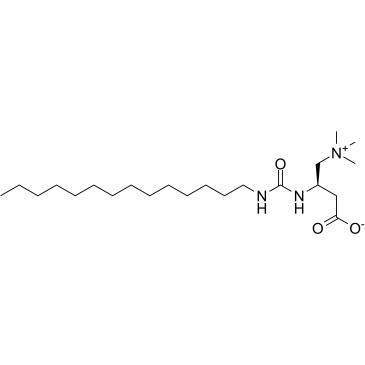
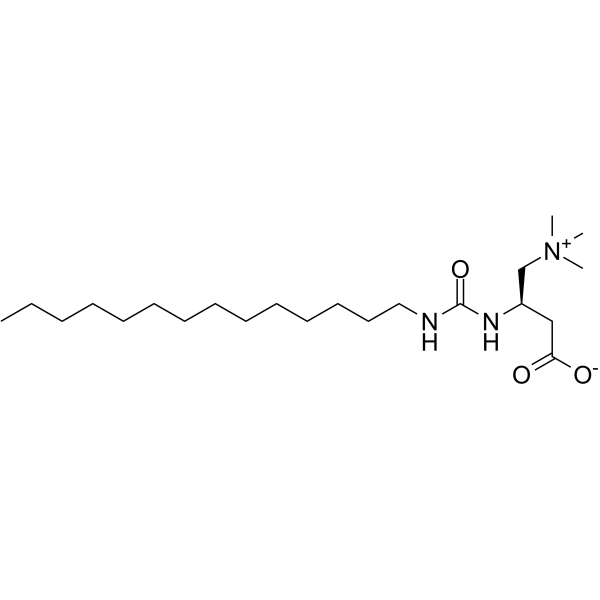
Teglicar 替格列卡
| 产品介绍 | Teglicar 是一种选择性、可逆性的肉碱棕榈酰转移酶1 (L-CPT1) 肝脏亚型抑制剂,具有口服活性,其 IC50 值为 0.68 μM, Ki 值为 0.36 μM。Teglicar 具有潜在的抗高血糖特性。Teglicar 可用于糖尿病和神经退行性疾病的研究,包括亨廷顿病(HD)。 | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 生物活性 | Teglicar is a selective and reversible orally active liver isoform of carnitine palmitoyl-transferase 1 (L-CPT1) inhibitor with an IC50 value of 0.68 μM and a Ki value of 0.36 μM. Teglicar has a potential antihyperglycemic propert. Teglicar can be used for the research of diabetes and neurodegenerative disease including Huntington's disease (HD). | ||||||||||||||||||||||||||||||||
| 体外研究 |
Teglicar has L-CPT1 inhibitory activity with an IC50 value of 0.68 μM and a Ki value of 0.36 μM. 西域 has not independently confirmed the accuracy of these methods. They are for reference only. | ||||||||||||||||||||||||||||||||
| 体内研究 |
Teglicar (oral, 80 mg/kg, once a day, for 30 days or infusion, 5.3 mg/kg/h, for 3 h) reduces the endogenous glucose production (262%) without affecting peripheral glucose utilization in SD rats. 西域 has not independently confirmed the accuracy of these methods. They are for reference only.
| ||||||||||||||||||||||||||||||||
| 体内研究 |
Teglicar (oral, 80 mg/kg, once a day, for 30 days or infusion, 5.3 mg/kg/h, for 3 h) reduces the endogenous glucose production (262%) without affecting peripheral glucose utilization in SD rats. 西域 has not independently confirmed the accuracy of these methods. They are for reference only.
| ||||||||||||||||||||||||||||||||
| 性状 | Solid | ||||||||||||||||||||||||||||||||
| 溶解性数据 |
In Vitro:
DMSO : 19 mg/mL (47.55 mM; Need ultrasonic) H2O : 10 mg/mL (25.02 mM; Need ultrasonic) 配制储备液
*
请根据产品在不同溶剂中的溶解度选择合适的溶剂配制储备液;一旦配成溶液,请分装保存,避免反复冻融造成的产品失效。 In Vivo:
请根据您的实验动物和给药方式选择适当的溶解方案。以下溶解方案都请先按照 In Vitro 方式配制澄清的储备液,再依次添加助溶剂:
——为保证实验结果的可靠性,澄清的储备液可以根据储存条件,适当保存;体内实验的工作液,建议您现用现配,当天使用;
以下溶剂前显示的百
| ||||||||||||||||||||||||||||||||
| 运输条件 | Room temperature in continental US; may vary elsewhere. | ||||||||||||||||||||||||||||||||
| 储存方式 |
| ||||||||||||||||||||||||||||||||
| 参考文献 |
|
 有竞争力的价格
有竞争力的价格匹配竞争对手的价格
 极速物流
极速物流效率为先
 技术支持
技术支持专业经验 贴心服务
 现货库存
现货库存50000+库存

 浙公网安备 33010802013016号
浙公网安备 33010802013016号
